
“`html
A protein situated in the incorrect area of a cell can lead to various ailments, including Alzheimer’s, cystic fibrosis, and cancer. However, there are approximately 70,000 distinct proteins and protein variations within an individual human cell, and as researchers can generally only analyze a few in one experiment, it becomes exceedingly expensive and time-consuming to manually determine the locations of proteins.
A novel generation of computational methods aims to simplify this procedure by utilizing machine-learning models that frequently employ datasets encompassing thousands of proteins and their respective locations, assessed across multiple cell lines. One of the most extensive such datasets is the Human Protein Atlas, which records the subcellular activities of over 13,000 proteins in more than 40 cell lines. Despite its vastness, the Human Protein Atlas has only examined about 0.25 percent of all potential combinations of proteins and cell lines within the database.
Currently, investigators from MIT, Harvard University, and the Broad Institute of MIT and Harvard have crafted a new computational strategy that can effectively explore the previously unexamined realms. Their approach is capable of predicting the location of any protein in any human cell line, even if neither the protein nor the cell has been previously tested.
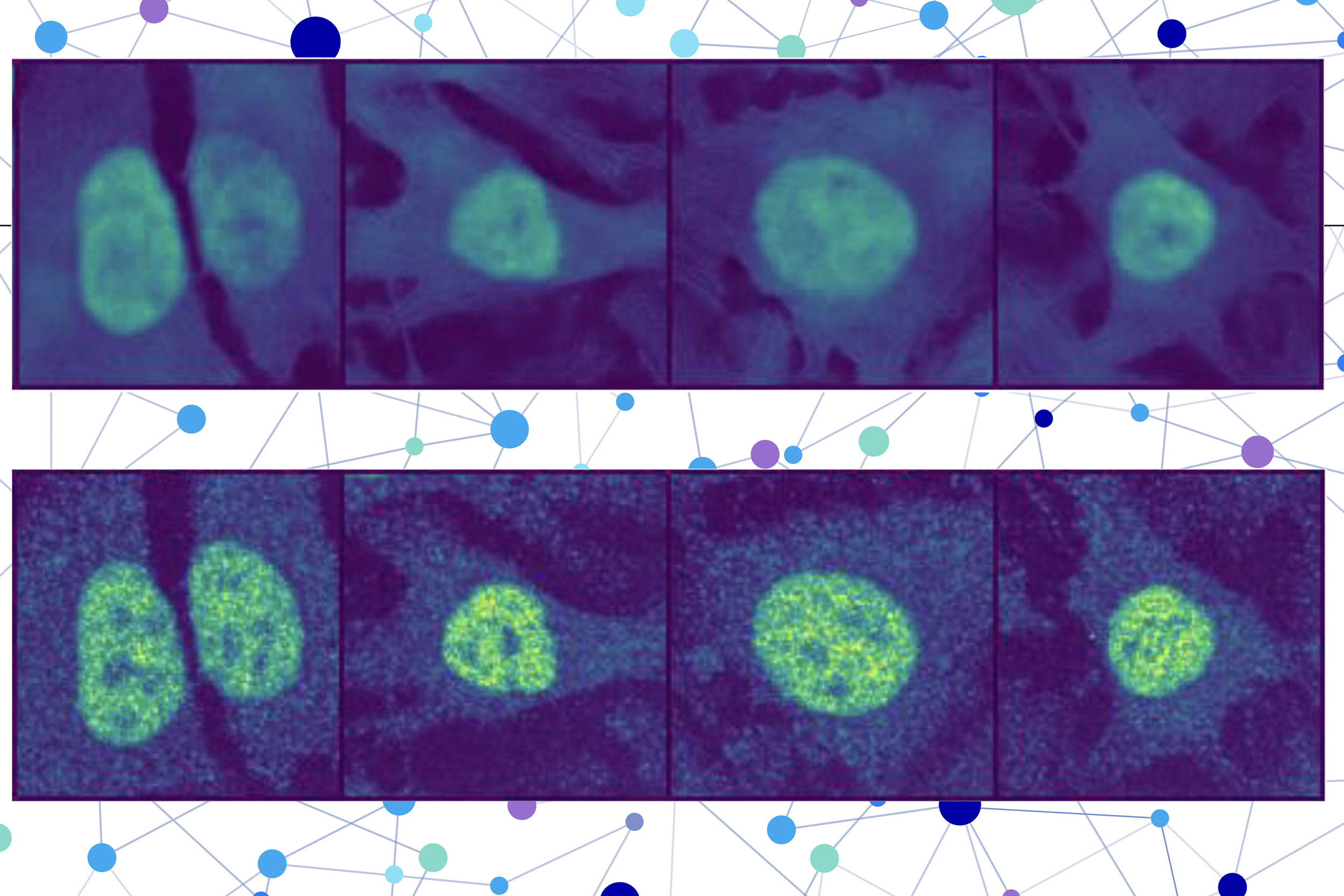
This technique advances beyond many AI-driven methods by localizing a protein at the single-cell level, rather than providing an averaged estimate across all cells of a specific type. For example, this single-cell localization could identify a protein’s position in a particular cancer cell post-treatment.
The researchers fused a protein language model with a specialized type of computer vision model to capture intricate details regarding a protein and its cell. Ultimately, the user receives a visual of a cell with a highlighted section indicating the model’s estimation of the protein’s position. Given that a protein’s localization reflects its functional status, this approach may aid researchers and clinicians in more effectively diagnosing diseases or identifying drug targets, while also allowing biologists to enhance their understanding of how complex biological processes relate to protein localization.
“You could perform these protein-localization experiments virtually without needing to engage with any lab equipment, potentially saving yourself months of effort. While you would still need to authenticate the predictions, this method could serve as a preliminary screening of what to investigate experimentally,” states Yitong Tseo, a graduate student in MIT’s Computational and Systems Biology program and co-lead author of a publication on this study.
Tseo is joined in the publication by co-lead author Xinyi Zhang, a graduate student in the Department of Electrical Engineering and Computer Science (EECS) and the Eric and Wendy Schmidt Center at the Broad Institute; Yunhao Bai of the Broad Institute; and senior authors Fei Chen, an assistant professor at Harvard and a member of the Broad Institute, and Caroline Uhler, the Andrew and Erna Viterbi Professor of Engineering in EECS and the MIT Institute for Data, Systems, and Society (IDSS), who also directs the Eric and Wendy Schmidt Center and is a researcher at MIT’s Laboratory for Information and Decision Systems (LIDS). This study has been published today in Nature Methods.
Collaborative Models
Numerous existing protein prediction models can solely forecast based on the protein and cell data they were trained with or are incapable of determining a protein’s location within a single cell.
To surmount these constraints, the researchers devised a two-part method for predicting the subcellular location of unseen proteins, termed PUPS.
The first component employs a protein sequence model to encapsulate the localization-determining attributes of a protein and its 3D structure derived from the chain of amino acids constituting it.
The second part integrates an image inpainting model, which is tasked with completing missing sections of an image. This computer vision model examines three stained images of a cell to extract information regarding the cell’s state, including its type, unique characteristics, and whether it is under stress.
PUPS merges the representations generated by each model to predict the protein’s location within a single cell, utilizing an image decoder to produce a highlighted image displaying the predicted location.
“Various cells within a cell line demonstrate different traits, and our model is adept at comprehending that nuance,” Tseo remarks.
A user inputs the sequence of amino acids that compose the protein along with three cell stain images — one for the nucleus, one for the microtubules, and one for the endoplasmic reticulum. Subsequently, PUPS manages the remainder.
A Deeper Comprehension
The researchers implemented several strategies during the training process to instruct PUPS on how to integrate information from each model in such a manner that it can make informed guesses regarding the protein’s location, even if it has not encountered that specific protein before.
For example, they allocated the model a secondary assignment during training: explicitly naming the localization compartment, such as the cell nucleus. This is conducted alongside the primary inpainting task to facilitate more effective learning.
A fitting analogy might be a teacher asking their students to sketch all the components of a flower in addition to labeling their names. This additional step was found to enhance the model’s overall understanding of the various cellular compartments.
Moreover, the simultaneous training of PUPS on proteins and cell lines aids it in cultivating a deeper insight into where proteins typically localize within the cellular image.
PUPS can even independently discern how different segments of a protein’s sequence contribute individually to its overall localization.
“Most other techniques usually necessitate that you have a stain of the protein first, meaning you’ve already observed it in your training data. Our methodology is exceptional in that it can generalize across proteins and cell lines concurrently,” Zhang notes.
By allowing PUPS to generalize to unseen proteins, it can capture localization alterations driven by unique protein mutations absent from the Human Protein Atlas.
The researchers confirmed that PUPS could predict the subcellular location of new proteins in unseen cell lines through laboratory experiments and by comparing the results. Furthermore, when assessed against a baseline AI method, PUPS displayed, on average, reduced prediction error across the proteins evaluated.
In the future, the researchers aspire to enhance PUPS so that the model can comprehend protein-protein interactions and make localization predictions for multiple proteins within a cell. In the long term, they aim to enable PUPS to make predictions in the context of living human tissue, instead of cultivated cells.
This research is supported by the Eric and Wendy Schmidt Center at the Broad Institute, the National Institutes of Health, the National Science Foundation, the Burroughs Welcome Fund, the Searle Scholars Foundation, the Harvard Stem Cell Institute, the Merkin Institute, the Office of Naval Research, and the Department of Energy.
“`