
A common, foundational aim among numerous chemistry scholars is the ability to forecast a molecule’s characteristics, such as its boiling or melting point. Once scientists can accurately make such predictions, they can advance their research, leading to breakthroughs in pharmaceuticals, materials, and more. Historically, however, conventional techniques for revealing these forecasts are linked to substantial costs — involving time and wear on equipment, as well as financial expenditure.
Enter a segment of artificial intelligence referred to as machine learning (ML). ML has alleviated the strain of predicting molecular properties to some extent, but the sophisticated tools that most efficiently accelerate the process — by deriving insights from existing data to generate quick forecasts for new molecules — demand a considerable level of programming knowledge from the user. This presents an accessibility challenge for many chemists, who may lack the necessary computational skillset to traverse the prediction pipeline.
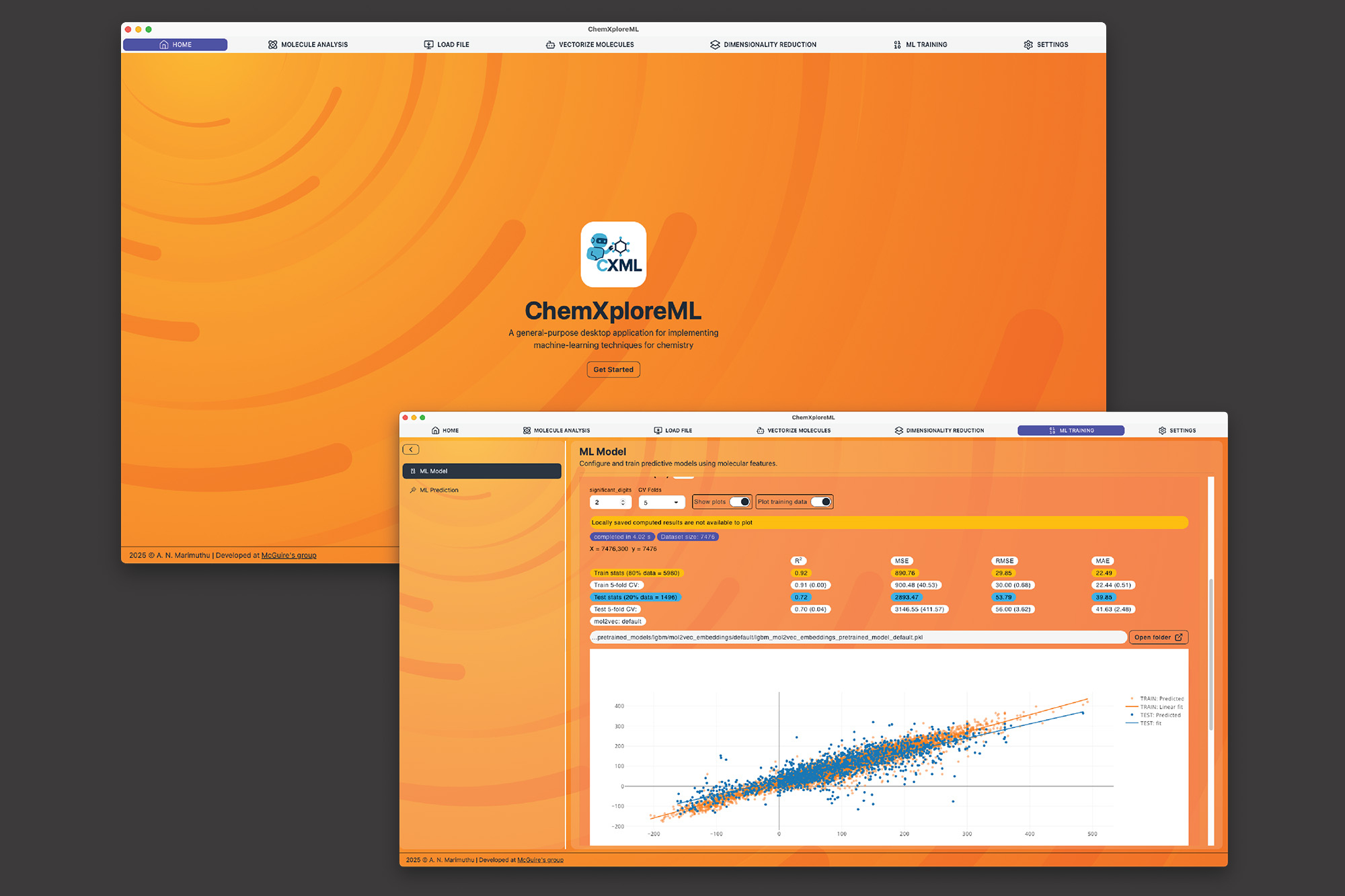
To tackle this issue, scientists in the McGuire Research Group at MIT have developed ChemXploreML, a user-friendly desktop application designed to assist chemists in making these essential predictions without the need for advanced programming abilities. Available at no cost, simple to download, and compatible with major platforms, this application is also engineered to function entirely offline, thereby safeguarding research data. This innovative technology is detailed in a recently published article in the Journal of Chemical Information and Modeling.
A particular obstacle in chemical machine learning is converting molecular architectures into a numeric format that computers can interpret. ChemXploreML automates this intricate procedure with robust, integrated “molecular embedders” that convert chemical structures into meaningful numerical vectors. Subsequently, the software employs cutting-edge algorithms to recognize patterns and predict molecular characteristics such as boiling and melting points, all via an intuitive, interactive graphical interface.
“The aim of ChemXploreML is to make machine learning accessible in the realm of chemical sciences,” states Aravindh Nivas Marimuthu, a postdoctoral researcher in the McGuire Group and principal author of the article. “By developing an intuitive, robust, and offline-capable desktop application, we are equipping chemists with state-of-the-art predictive modeling tools, irrespective of their programming expertise. This endeavor not only quickens the discovery of new drugs and materials by streamlining the screening process, but its adaptable framework also paves the way for future innovations.”
ChemXploreML is intended to progress over time, allowing new techniques and algorithms to be effortlessly integrated into the application, ensuring that researchers have continual access to the latest methodologies. The application underwent testing for five crucial molecular properties of organic compounds — melting point, boiling point, vapor pressure, critical temperature, and critical pressure — achieving high precision scores of up to 93 percent for critical temperature. The researchers also showed that a novel, more compact method of representing molecules (VICGAE) was nearly as precise as traditional methods, such as Mol2Vec, yet was up to 10 times quicker.
“We envision a future where any researcher can effortlessly customize and employ machine learning to address distinct challenges, ranging from creating sustainable materials to delving into the intricate chemistry of interstellar space,” remarks Marimuthu. He is joined on the paper by senior author and Class of 1943 Career Development Assistant Professor of Chemistry Brett McGuire.