
All biological activities rely on how various proteins engage with one another. Protein-protein engagements enable processes from the transcription of DNA and regulation of cell proliferation to advanced functions in intricate organisms.
Nevertheless, much remains ambiguous regarding how these functions are coordinated on the molecular scale and how proteins associate with one another — whether with distinct proteins or with their own copies.
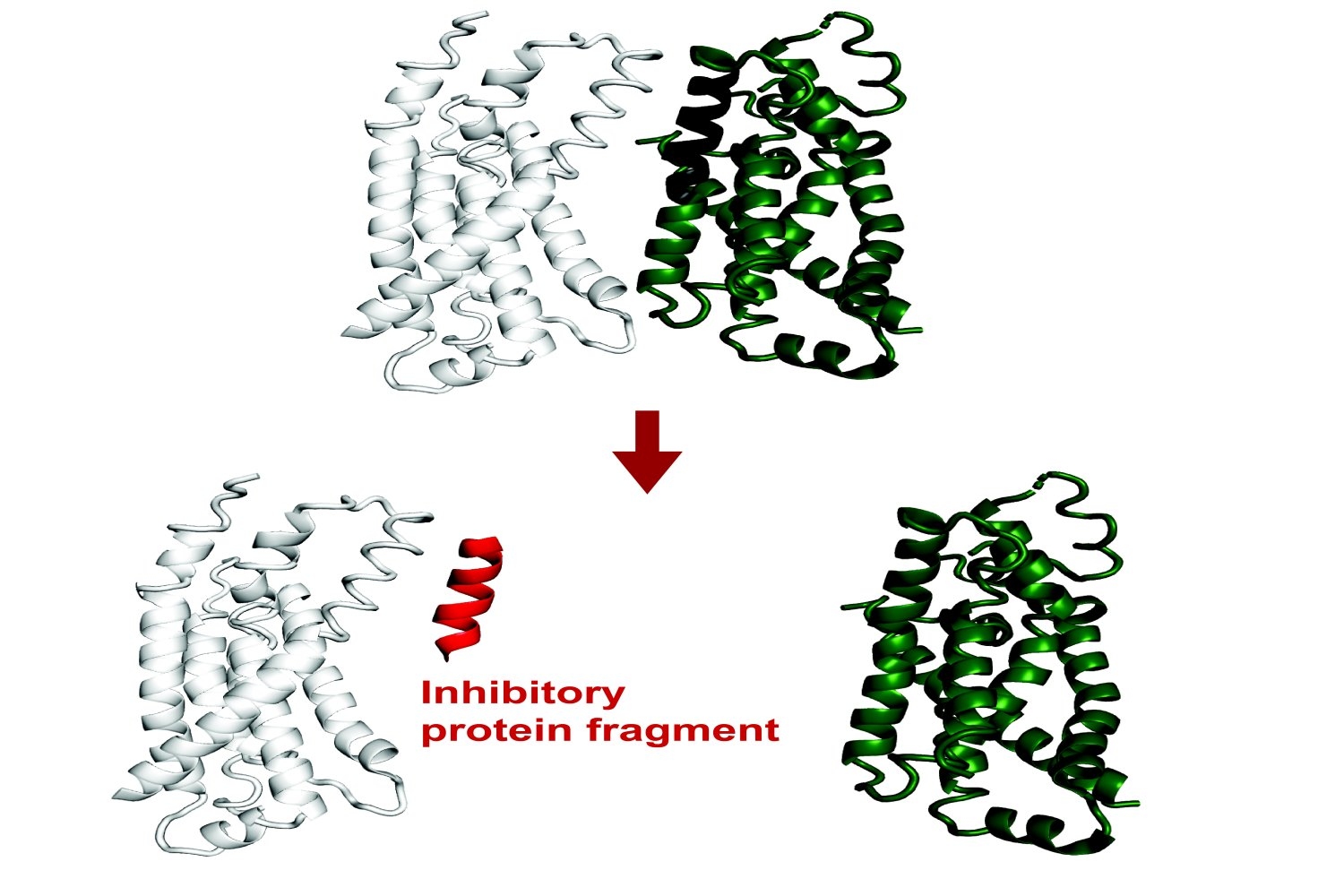
Recent research has uncovered that small protein segments possess considerable functional capabilities. Despite being incomplete fragments, short sequences of amino acids can still connect to the interfaces of a target protein, mimicking natural interactions. Through this mechanism, they can modify that protein’s functionality or interfere with its interactions with other proteins.
Consequently, protein fragments could enhance both fundamental studies on protein interactions and cellular functions, and may hold promising therapeutic prospects.
Recently published in Proceedings of the National Academy of Sciences, an innovative technique developed in the Department of Biology builds upon pre-existing artificial intelligence frameworks to computationally forecast protein fragments capable of binding to and obstructing full-length proteins in E. coli. Theoretically, this tool could pave the way for genetically encodable inhibitors against any protein.
The research was conducted in the laboratory of associate professor of biology and Howard Hughes Medical Institute researcher Gene-Wei Li in partnership with the lab of Jay A. Stein (1968) Professor of Biology, professor of biological engineering, and department chair Amy Keating.
Utilizing machine learning
The software, referred to as FragFold, employs AlphaFold, an AI model that has enabled remarkable progress in biology over the past few years due to its proficiency in predicting protein folding and interactions.
The project’s aim was to forecast fragment inhibitors, representing a novel utilization of AlphaFold. The researchers experimentally verified that over half of FragFold’s forecasts regarding binding or inhibition were accurate, even when they lacked prior structural data on the mechanisms of those interactions.
“Our findings indicate that this is a generalized method for identifying binding modes likely to inhibit protein activity, encompassing novel protein targets, and these predictions can serve as a foundation for additional experiments,” asserts co-first and corresponding author Andrew Savinov, a postdoctoral researcher in the Li Lab. “We can effectively apply this towards proteins without known functions, interactions, or even structures, and we can instill some confidence in these models we’re developing.”
One instance is FtsZ, a protein essential for cell division. It is extensively studied but harbors a region that is intrinsically disordered and, as such, poses particularly challenging obstacles for examination. Disordered proteins are dynamic, and their functional interactions are likely transient — happening so momentarily that contemporary structural biology tools fail to capture a single structure or interaction.
The researchers utilized FragFold to investigate the behavior of fragments of FtsZ, including those from the intrinsically disordered region, to uncover several new binding associations with various proteins. This advancement in understanding confirms and broadens previous experiments that assessed FtsZ’s biological functions.
This advancement is noteworthy mainly because it was achieved without determining the structure of the disordered region and because it showcases the potential strength of FragFold.
“This illustrates how AlphaFold is fundamentally transforming our approach to studying molecular and cellular biology,” Keating states. “Innovative applications of AI methodologies, such as our work on FragFold, unveil unexpected possibilities and new research pathways.”
Inhibition, and beyond
The researchers achieved these predictions by computationally fragmenting each protein and then modeling how those fragments would bind to interaction partners deemed relevant.
They juxtaposed the maps of predicted binding across the entire sequence with the effects of those same fragments in live cells, assessed using high-throughput experimental measures in which millions of cells each produce one type of protein fragment.
AlphaFold employs co-evolutionary information to forecast folding, typically analyzing the evolutionary background of proteins using a method known as multiple sequence alignments for every single prediction run. The MSAs play a critical role, but pose a bottleneck for large-scale forecasts — consuming an excessive amount of time and computational resources.
For FragFold, the researchers opted to pre-calculate the MSA for a full-length protein once and utilized that output to inform predictions for each fragment of that full-length protein.
Savinov, along with Keating Lab alumnus Sebastian Swanson PhD ’23, predicted inhibitory fragments of a varied set of proteins beyond FtsZ. Among the pairings they examined was a complex between lipopolysaccharide transport proteins LptF and LptG. A protein fragment of LptG inhibited this interaction, presumably obstructing the transport of lipopolysaccharide, an essential component of the E. coli outer cell membrane critical for cellular integrity.
“The major revelation was our ability to predict binding with such high precision and, in fact, often to foresee bindings that correspond to inhibition,” Savinov remarks. “For every protein we’ve investigated, we’ve successfully identified inhibitors.”
The researchers primarily concentrated on protein fragments as inhibitors because determining whether a fragment could impede a vital function in cells is a relatively straightforward outcome to assess systematically. Looking ahead, Savinov is also keen on investigating fragment functions beyond inhibition, such as fragments capable of stabilizing the proteins they bind to, enhancing or modifying their activity, or initiating protein degradation.
Design, in principle
This research serves as a foundational step for developing a systematic comprehension of cellular design principles and what characteristics deep-learning models may utilize to make accurate predictions.
“There’s a broader, more expansive goal we’re aspiring toward,” Savinov mentions. “Now that we can forecast them, can we leverage the data we obtain from predictions and experiments to extract the pertinent features to discern what AlphaFold has truly learned about what constitutes an effective inhibitor?”
Savinov and collaborators also delved deeper into the binding of protein fragments, probing additional protein interactions and mutating specific residues to observe how those alterations affect interactions between the fragment and its target.
Experimentally investigating the behavior of thousands of mutated fragments within cells, an approach identified as deep mutational scanning, uncovered crucial amino acids responsible for inhibition. In several instances, the mutated fragments exhibited even greater inhibitory potency than their natural, full-length counterparts.
“Unlike previous techniques, we are not constrained to identifying fragments from experimental structural data,” states Swanson. “The core strength of this work stems from the synergy between high-throughput experimental inhibition data and the predicted structural models: the experimental data directs us to fragments that are particularly notable, while the structural models anticipated by FragFold furnish a specific, testable hypothesis regarding how the fragments operate on a molecular scale.”
Savinov is enthusiastic about the prospects of this approach and its myriad possibilities.
“By developing compact, genetically encodable binders, FragFold opens up a extensive range of opportunities to manipulate protein activity,” Li concurs. “We can envision delivering functionalized fragments that can modify native proteins, alter their subcellular localization, and even reprogram them to create innovative tools for exploring cell biology and addressing diseases.”