Proteins serve as the essential engines that maintain the functionality of our cells, with numerous varieties of proteins performing distinct duties within them. It has been established for a long time that a protein’s configuration dictates its functionality. More recently, scientists have begun to recognize that a protein’s positioning is equally essential to its performance. Cells contain numerous compartments that help organize their various inhabitants. In addition to the familiar organelles depicted in biology textbooks, these areas also encompass a range of dynamic, membrane-less compartments that cluster certain molecules together to execute collaborative tasks. Understanding where a specific protein localizes, and its co-localization partners, can thus aid in the deeper comprehension of that protein’s role in both healthy and diseased cells, but researchers have struggled to find a systematic approach to predict this information.
On the other hand, protein structure has been thoroughly examined for more than fifty years, culminating in the development of the artificial intelligence tool AlphaFold, capable of forecasting protein structure from a protein’s amino acid sequence, the linear arrangement of building blocks that folds to establish its shape. AlphaFold and similar models have emerged as widely employed resources in research.
Additionally, proteins possess segments of amino acids that do not form a stable structure, yet are crucial for facilitating proteins’ entry into dynamic compartments within the cell. MIT Professor Richard Young and his team questioned whether the information in those segments could be leveraged to forecast protein localization similarly to how other sections are utilized to predict structure. Other scientists have identified several protein sequences that indicate protein localization, with some beginning the creation of predictive models for protein localization. However, it remained unclear if a protein’s location within any dynamic compartment could be anticipated based on its sequence, nor did researchers have a tool akin to AlphaFold for localization predictions.
Now, Young, who is also part of the Whitehead Institute for Biological Research; Young lab postdoctoral fellow Henry Kilgore; Regina Barzilay, the School of Engineering Distinguished Professor for AI and Health in MIT’s Department of Electrical Engineering and Computer Science and principal investigator in the Computer Science and Artificial Intelligence Laboratory (CSAIL); along with colleagues have developed such a model, which they have named ProtGPS. In a publication released on Feb. 6 in the journal Science, featuring first authors Kilgore and Barzilay lab graduate students Itamar Chinn, Peter Mikhael, and Ilan Mitnikov, the interdisciplinary team introduces their model. The researchers demonstrate that ProtGPS can accurately predict to which of 12 known types of compartments a protein will localize, along with whether a mutation linked to a disease will alter that localization. Furthermore, the research group created a generative algorithm capable of designing novel proteins intended to localize in specific compartments.
“I aspire that this marks a foundational step towards a robust platform that empowers researchers studying proteins to further their investigations,” Young remarks. “Moreover, it may enhance our understanding of how humans evolve into the complex organisms they are, how mutations disrupt those natural processes, and how to formulate therapeutic hypotheses and develop medications to address cellular dysfunction.”
The researchers also confirmed numerous predictions made by the model through experimental testing in cellular settings.
“I was genuinely thrilled to transition from computational design to testing these outcomes in the laboratory,” Barzilay states. “There are numerous fascinating publications in the AI domain, but 99.9 percent of those never undergo validation in real-world systems. Thanks to our partnership with the Young lab, we could test our predictions and truly ascertain how well our algorithm performs.”
Creating the model
The group trained and evaluated ProtGPS using two sets of proteins with established localizations. They found that it could accurately predict the eventual locations of proteins with high precision. The researchers additionally assessed how well ProtGPS could forecast alterations in protein localization due to disease-associated mutations within a protein. Numerous mutations — changes to the genetic sequence that correlates with a gene and its protein — have been identified as contributors to or instigators of disease based on correlation studies, yet the mechanisms through which mutations cause disease manifestations remain elusive.
Understanding the process by which a mutation contributes to illness is crucial as it enables researchers to develop treatments aimed at rectifying that mechanism, thus preventing or managing the disease. Young and collaborators conjectured that several mutations associated with diseases might lead to illness through alterations in protein localization. For instance, a mutation may prevent a protein from associating with a compartment containing vital partners.
They examined this hypothesis by inputting over 200,000 proteins with disease-related mutations into ProtGPS, subsequently asking it to both predict where those mutated proteins would localize and assess the extent to which its predictions varied between the normal and mutated forms. A significant alteration in prediction signifies a probable change in localization.
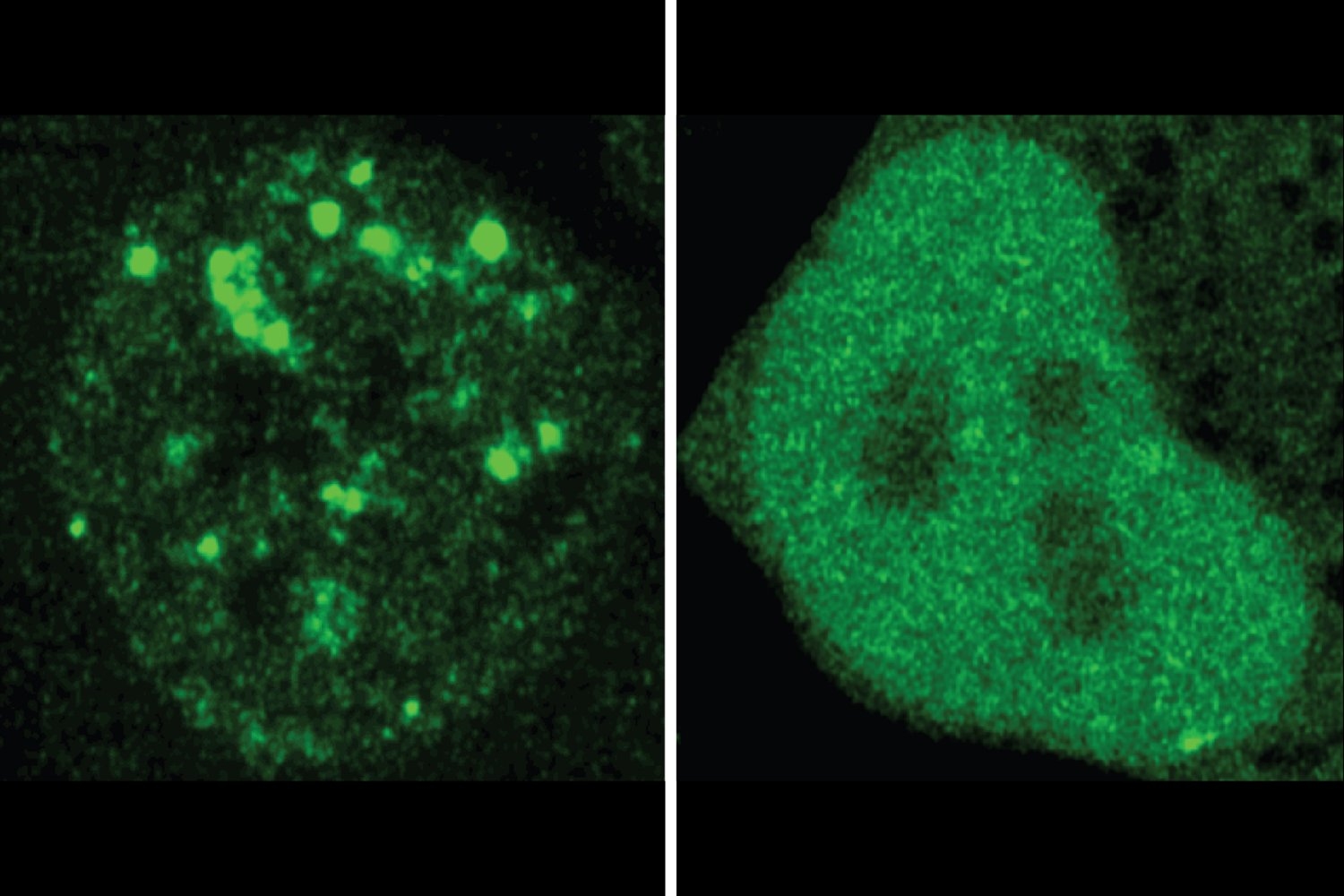
The researchers discovered numerous instances wherein a disease-related mutation appeared to modify a protein’s localization. They examined 20 such cases in cells, utilizing fluorescence to compare the cellular locations of normal proteins and their mutated counterparts. The experimental results validated ProtGPS’s predictions. Collectively, the findings support the researchers’ hypothesis that mis-localization may be an often-overlooked mechanism of disease, highlighting the utility of ProtGPS as a resource for understanding disease processes and uncovering new therapeutic opportunities.
“The cell is an immensely intricate system, replete with diverse components and complex interaction networks,” Mitnikov notes. “It’s incredibly intriguing to consider that through this method, we can manipulate the system, observe the resulting changes, and consequently drive the discovery of cellular mechanisms, or even formulate therapeutics based on that.”
The researchers hope that others will begin to utilize ProtGPS in a manner similar to how predictive structural models like AlphaFold are employed, pushing forward various initiatives concerning protein function, dysfunction, and disease.
Advancing from prediction to novel generation
Although the researchers were enthusiastic about the potential applications of their prediction model, they also aimed for it to exceed mere localization predictions of existing proteins, allowing them to design entirely new proteins. The objective was for the model to create completely novel amino acid sequences that, once synthesized in a cell, would localize to a specified area. Creating a novel protein that can indeed fulfill a role — in this instance, the function of localizing to a particular cellular compartment — is remarkably challenging. To enhance their model’s likelihood of success, the researchers limited their algorithm to design proteins analogous to those found in nature. This approach is frequently employed in drug design for logical reasons; nature has had billions of years to discover which protein sequences are effective and which are not.
Due to the collaboration with the Young lab, the machine learning team was able to verify whether their protein generator functioned effectively. The model yielded positive results. In one iteration, it generated 10 proteins intended to localize to the nucleolus. Upon testing these proteins in cellular environments, researchers found that four of them strongly localized to the nucleolus, while others exhibited slight preferences toward that location as well.
“The collaboration between our labs has proven to be extremely productive for all parties involved,” Mikhael states. “We’ve learned to communicate in each other’s terms, particularly regarding cellular functions, and by having the opportunity to experimentally validate our model, we’ve discerned what adjustments are necessary to enhance its efficacy.”
Generating functional proteins in this manner could considerably enhance researchers’ capabilities to innovate therapies. For instance, if a medication needs to interact with a target situated within a particular compartment, then researchers could employ this model to devise a drug that also localizes there. This should enhance the drug’s effectiveness and minimize side effects, as it would spend more time engaging with its target and less time interacting with extraneous molecules, thereby decreasing off-target interactions.
The machine learning team members are excited about the potential of utilizing insights gained from this collaboration to design novel proteins encompassing other functions beyond localization, thereby broadening the scope of therapeutic design and other applications.
“A multitude of studies demonstrate the capability to create proteins that can be expressed in a cell, but not necessarily those that exhibit specific functions,” Chinn remarks. “We’ve accomplished functional protein design with a relatively high success rate compared to other generative models. This is genuinely thrilling for us, and something we aim to expand upon.”
All researchers involved perceive ProtGPS as an exhilarating starting point. They anticipate that their tool will facilitate further exploration of the significance of localization in protein function and mis-localization in diseases. Additionally, they are keen on broadening the model’s localization predictions to encompass more types of compartments, examining more therapeutic hypotheses, and developing increasingly functional proteins for therapeutic or other uses.
“Now that we are aware that this protein code for localization exists, and that machine learning models can decipher that code and even generate functional proteins using its principles, it opens a myriad of potential studies and practical applications,” Kilgore asserts.